Longevity Journal Club 2022 Edition

Curriculum Creator
Martin Borch Jensen
Presenters
Divya Cohen
Tara Mei
Lada Nuzhna
Kush Sharma
Madison Ueland
Introduction
Goals
The goals of the Longevity Journal Club are threefold:
Understand the history of longevity research and how the field developed over time
Be able to critically appraise past and current research
Develop taste to understand areas of novelty and/or where further research could be fruitful
In order to do this, landmark papers in the field were selected that explored the earliest history as well as the various subfields of longevity research. This list is not exhaustive of all areas. It covers areas or papers Martin Borch Jensen believes are some of the most interesting and important to understand for newcomers to the field. Over time, new papers and new subfields will be added to the curriculum.
This document hopes to share our learnings openly so others can gain from these presentations and discussions.
Who is Involved
Papers were divided among the 2021-2022 Longevity Apprentices who were responsible for presenting its findings. Each paper is linked to the associated powerpoint presentation. The discussion summary sections at the end of each paper tried to capture what was discussed in each of the live sessions.
Structure
The curriculum starts from 1983 when the field first realized there is genetics at least partially governing longevity. In order to make the most of the curriculum, we recommend you either start from the beginning so you too can develop an understanding of how the field has evolved, but you could also skip to sections of greatest interest. Each topic includes the motivation for curriculum inclusion, to set context for why this is considered an important paper in the field or further expands our collective knowledge. To get a quick understanding of what each paper showed, read the summary.
Feedback or Comments
If you’d like to make comments, provide feedback and/or suggest other papers to add to the curriculum, please email martin@norn.group.
- Coupling of late-life reproduction with lifespan (1983)
- The first discovery of longevity genes (1989-1993)
- Sirtuins/resveratrol controversy (1999-2008ish)
- NIA Intervention Testing Program (2000-...)
- Mitochondria in aging (2004)
- Heterochronic parabiosis (2005)
- Nutrient Sensing
- Late-life rapamycin intervention (2009)
- Caloric restriction in primates extends/doesn’t extend lifespan (2009-2012)
- Clearance of senescent cells improves health (2011)
- The (over)influential Hallmarks of Aging framework (2013)
- Horvath methylation clock (2013)
- What is immune aging? (2013-)
- Partial rejuvenation in vivo helps progeria mice (2016)
- Human GWAS studies (2019)
- Measuring aging in humans (2019)
- Statistics of Biology
- Summary of longevity intervention data in humans
Coupling of late-life reproduction with lifespan (1983)

Paper(s): Rose M. R. (1984). Laboratory Evolution of Postponed Senescence in Drosophila Melanogaster. Evolution; international journal of organic evolution, 38(5), 1004–1010.
Motivation for including: Even before anyone had directly shown that genes can regulate lifespan, Michael Rose’s experiments showed that lifespan could be genetically selected for, and that reproduction and lifespan are not intrinsically opposed. These studies also led to the largest recorded lifespan increase in Drosophila (3-4x over controls).
Journal Club Presenter: Divya Cohen
Slides: Link
Presentation date: 1/16/2022
Summary
Confirmed Earlier Studies’ Hypothesis
Longevity increased over generations by propagating progeny from later ages (earlier studies only confirmed this for females)
Fecundity is redistributed to later for longer living populations while viability is preserved.
Stringent controls are important for replicability of study.
The method for the experiment and controls included:
Ten experimental populations of Drosophila melanogaster
were derived from single base (B) generation
Five of these populations termed B were maintained for 50 generations
The other five were maintained in the same conditions as B population except eggs collected for the next generation were progressively postponed
The assays measured included a) Fecundity, b) Viability, and c) Longevity
A limitation of the study is its lack of insight into the biological reason for the difference in longevity
Since then, these flies have continued to be bred at progressively longer reproductive intervals and are owned by a company called Genescient, that is no longer actively developing new insights from them.
The life extension population at present day now have a ~180 day life span (~3.5x that of controls)
Background / Context
In 1983 the predominant theory was the Evolution Theory of Senescence which proclaims the force of natural selection declines with age where senescence is defined by decline in age-specific fitness-components after the onset of reproductive maturity. There are two derived corollaries, a) earlier age of reproduction accelerates senescence and b) later age of reproduction postpones senescence. These theories were tested by several labs at time, especially Lint who could not reproduce the results of noted due to technical artifacts such as inbreeding, genetic disequilibrium, and inadequate controls. The purpose of the Rose study was to reproduce the experiments from previous studies but with adequate controls.
Discussion Summary
In addition to the facts shown in the presentation, for this subject, it’s important to understand the narrative progression of events and the individuals involved.
There were questions about why isn’t longevity selected for in the wild? One explanation is the rate of external mortality such as due to predators or accidents etc could be constant but with earlier reproduction timelines, the genes for longevity do not become concentrated as offsprings have more offspring. This could also explain why females live longer across many species, as males across many species have higher rates of extrinsic mortality (link).
The evolutionary theory of senescence is consistent with the disposable soma theory (link) which states that organisms age due to an evolutionary trade-off between longevity and reproduction.
Since this study, the Drosophila melanogaster flies from this experiment have continued to be bred at progressively longer reproduction windows. A more recent (2013) genomic study of an offshoot fly population showed a RNAseq analysis with limited new insights. It showed transcriptional responses covering over 19% of the genome with genes affecting proteolysis, metabolism, oxidative phosphorylation, and mitochondrial function, protein synthesis, immunity, defense responses, and the detoxification of xenobiotic substances.
The first discovery of longevity genes (1989-1993)

Paper(s): Kenyon C. (2011). The first long-lived mutants: discovery of the insulin/IGF-1 pathway for ageing. Philosophical transactions of the Royal Society of London. Series B, Biological sciences, 366(1561), 9–16.
Motivation for including: Before these studies, it was not known whether individual genes could affect the aging process. daf-2 mutants have ~doubled lifespan, decisively proving that aging is regulated genetically.
Journal Club Presenter: Martin Borch Jensen
No slides
Presentation date: 12/19/2021
Summary
Genetic studies on the small roundworm Caenorhabditis elegans established the first genetic pathway sufficient for significant life extension (insulin/IGF-1 signaling). Later studies have validated lifespan and delayed onset of aging diseases in multiple species. This paper summarizes how this discovery was made, and touches on the (very nascent) state of the aging field at the time.
The Johnson and later Kenyon papers were the first examples of single genes regulating lifespan, proving this is possible and thus opening the possibility for more research.
Prior to these papers, the 'aging' field was focused on physiology, 'damage accumulation/wear and year', and was largely theoretical. This marks a turning point towards molecular biology and genetics of aging. To put the ‘historical’ context differently: The molecular study of aging has only been studied since Nirvana.
This paper emphasizes a new focus on "regulatory" genes as opposed to enzymatic.
As it happens, this pathway (IIS, FoxO) is still one of two (alongside mTOR) that have most robustly extended lifespan (but we still don't truly understand how, in the sense of ‘what happens that prevents mortality’).
Discussion Summary
The historical summary, by just one of the key players, is surprisingly nuanced and fair. Minimal if any ‘revisionist history’ to amplify own role.
Thought experiment: Could we just target two main proteins (FOXO and mTOR). For example, use rapamycin.
Possible to do so but rapamycin is off patent. Thus startups are doing rapalogues.
Could consider raising philanthropic funds to take off patent rapamycin into clinical trials. Shows use of an arsenal of existing drugs.
FOXO is a transcription factor, more difficult to target with traditional drug modalities. Also, there are multiple FOXOs in humans (worms have just 1).
(See additional discussion of rapamycin in Late-life rapamycin intervention (2009))
Sirtuins/resveratrol controversy (1999-2008ish)

Paper(s): Summary - Garber, K. A mid-life crisis for aging theory. Nat Biotechnol 26, 371–374 (2008), Primary data refutations Burnett, C., Valentini, S., Cabreiro, F., Goss, M., Somogyvári, M., Piper, M. D., Hoddinott, M., Sutphin, G. L., Leko, V., McElwee, J. J., Vazquez-Manrique, R. P., Orfila, A. M., Ackerman, D., Au, C., Vinti, G., Riesen, M., Howard, K., Neri, C., Bedalov, A., Kaeberlein, M., … Gems, D. (2011). Absence of effects of Sir2 overexpression on lifespan in C. elegans and Drosophila. Nature, 477(7365), 482–485. and Beher, D., Wu, J., Cumine, S., Kim, K. W., Lu, S. C., Atangan, L., & Wang, M. (2009). Resveratrol is not a direct activator of SIRT1 enzyme activity. Chemical biology & drug design, 74(6), 619–624.
Motivation for including: Sirtuins were one of the first classes of proposed pro-longevity genes, with sirtuin activators promoted as anti-aging drugs. The genes as well as the drugs were subject to enormous controversy, and are a good case study of conflicting claims and individual interests in the aging field.
Journal Club Presenter: Kush Sharma
Slides: Link
Presentation date: 1/23/2022
Summary
Sirtuins (Sir2 in yeast and SIRT1 in mammals) are conserved genes, proposed in the 1990s and 2000s to be pro-longevity genes, with resveratrol (a small molecule found in red wine) proposed as a sirtuin activator, and therefore anti-aging drug
Early studies found that sirtuin overexpression/activation by resveratrol extended lifespan, however, these studies later failed to replicate or were otherwise brought into question
Many main players in the back-and-forth scientific controversy came from the same lab (Guarente), and went on to publish contradictory and competing research on the effects of sirtuins and resveratrol
Sirtris, the biotech commercializing sirtuin activators, was founded on research by David Sinclair that received lots of popular press attention and capitalized well on that attention in its sale to GSK for $720M
In retrospect, sirtuin activators have small effect sizes, significant negative evidence, and from the perspective of a drug developer, likely aren’t worth attention as a pro-longevity drug candidate
Mammals have seven sirtuins (SIRT1-SIRT7); although SIRT1 is the subject of discussion here and the most studied, SIRT6 overexpression has shown lifespan extension in mice
Background / Context
Sirtuins were among the earliest proposed conserved pro-longevity genes. They were initially discovered to increase lifespan when overexpressed in yeast. It was then proposed that they intermediated the lifespan-elongating effects of calorie restriction (CR). Overexpression of Sir2 homologs in higher organisms was shown to extend lifespan, and resveratrol, a proposed Sir2 activator, was pushed as an anti-aging compound. Sirtuins were brought to the forefront of aging research, with substantial attention dedicated to them for several years. A biotech, Sirtris, was spun up around the research and sold in a high-profile acquisition to GSK. As time went on, some key experiments failed to replicate. Eventually, the Sirtris acquisition was shuttered and sirtuin activator research was shelved at GSK. Sirtuin research became less dominant over time.
Discussion Summary
In addition to the facts shown in the presentation, for this subject, it’s important to understand the narrative progression of events and the individuals involved.
Most of the main players came from the same lab - Matt Kaeberlein, David Sinclair, and Brian Kennedy all spent overlapping years in Lenny Guarente’s MIT lab. Matt and Brian started labs on the West Coast, while David stayed close by at Harvard. David and Lenny continued publishing together and following somewhat similar research agendas for a few years, while Brian and Matt diverged, and published some of the main anti-sirtuin / resveratrol evidence. Brian and Matt also went on to uncover the importance of mTOR for longevity and found that it was one of the intermediary mechanisms for calorie restriction (CR), while David and Lenny were pushing SIRT1 as the CR mechanism. Other main players include Linda Partridge (University College London, anti-sirtuin), who, with Kaeberlein, failed to reproduce Sinclair’s Sir2 overexpression in flies and worms experiment, and Rafael de Cabo (National Institute on Aging, pro-sirtuin) who, with Sinclair, ran early resveratrol experiments in mice showing phenotypic improvements.
Sirtris Pharmaceuticals was the main corporate promoter of resveratrol/sirtuin activators and by extension the sirtuin<>longevity hypothesis. It was founded by David Sinclair, advised by Lenny Guarente, and based on IP from both labs. It received lots of popular press attention - the narrative that a compound from red wine (resveratrol) was anti-aging was understandably catchy. It IPOed 6 months after a high-profile publication from Sinclair and the NIA that resveratrol extended lifespan in a high-fat, high-calorie mouse model. It was later acquired by GSK. Its compounds, resveratrol and other proposed sirtuin activators, were tested for a number of years. Eventually, the subdivision was shut down and the assets were shelved due to varying lacks of safety, efficacy, and patentability.
Another question that was discussed: if the effect size is so small, and there’s both positive and negative evidence, is it worth ‘getting down to the ground truth’ and figuring out what’s going on, or should we just move on? If a drug developer was looking at the available data and didn’t have a strong human genetic argument for the importance of sirtuins, they likely wouldn’t run a human longevity trial with these compounds / for sirtuins. There may be arguments for other diseases, e.g. metabolic diseases like Type 2 Diabetes, since SIRT1 is involved in metabolism. GSK ran Phase II trials on Type 2 Diabetes for another Sirtris sirtuin activator, SRT2104.
Finally, the question of the other six sirtuins other than SIRT1. SIRT1 has received much of the attention, particularly in the 2000s/2010s, but SIRT6 has also shown lifespan extension effects. This won’t be considered in-depth here, but overexpression of SIRT6 in mice increases male, but not female lifespan by 10-15%.
NIA Intervention Testing Program (2000-...)

Program Link: https://www.nia.nih.gov/research/dab/interventions-testing-program-itp
Motivation for including: An effort to robustly reproduce and find new lifespan regulators. Three sites with coordinated protocols, outbred mouse populations, both sexes and cohorts big enough to detect ~10% effects. Considered the gold standard for longevity intervention testing.
Journal Club Presenter: Madison Ueland
Slides: Link
Presentation date: 1/30/2022
Takeaways
The NIA’s Interventions Testing Program (ITP), established in 2003, is the gold standard for lifespan interventions in mice.
The ITP tests a variety of single drug or dietary interventions in genetically heterogeneous mice across three different locations.
Compounds are tested in male and female mice, and most effects have been found to be sex-specific: the majority of the positive results (including aspirin, 17-alpha-estradiol, NDGA, protandim, canagliflozin) only extended lifespan of male mice.
While most compounds are only tested once (in 4-month old mice, at the dose recommended by the sponsor), several drugs have been tested in older mice. Notably rapamycin works equally as well late in life (20-month old mice), as does 17-alpha-estradiol in late middle age mice (16-month old mice).
Negative results include resveratrol, metformin and NR, but negative results come with a couple caveats: Phase I of the ITP primarily monitors lifespan so may miss healthspan extending interventions, and most interventions are only tested in a single dose.
Background
The ITP was conceived at a 1999 NIA workshop, when lifespan extending interventions testing were sporadic and compromised by the use of small sample sizes, limited biomarker assessment and infrequent publishing of negative results. At the time, caloric restriction (CR) was the only intervention validated to extend lifespan in mammals. The ITP set out to rigorously validate claims about single drug interventions and identify promising interventions in animals that might lead to clinical trials in humans.
Discussion Summary
There were a range of considerations made when choosing the animal model for the ITP (see slides); our discussion focused on genetic heterogeneity and causes of death.
The standard mouse strain used in most labs is Black 6 (C57/BL6), an inbred strain that was the first mouse strain (and the second mammal) to have its entire genome published. Black 6 mice are popular largely due to inertia and the scientific lore that inbred strains should display less phenotypic variability than outbred mice. In theory, reduced variation between mice enables the use of smaller sample sizes for a given level of statistical power, though this argument that using inbred strains increases replicability has been called into question.
Given the downsides of inbred mice (including congenital defects and smaller litter sizes), the ITP set out to achieve reproducibility without genetic homogeneity. The ITP uses UM-HET3, a four-strain cross developed by Richard Miller that breeds two different F1 hybrids, forming heterogeneous mice that each share a random 50% of their DNA (see sketch).
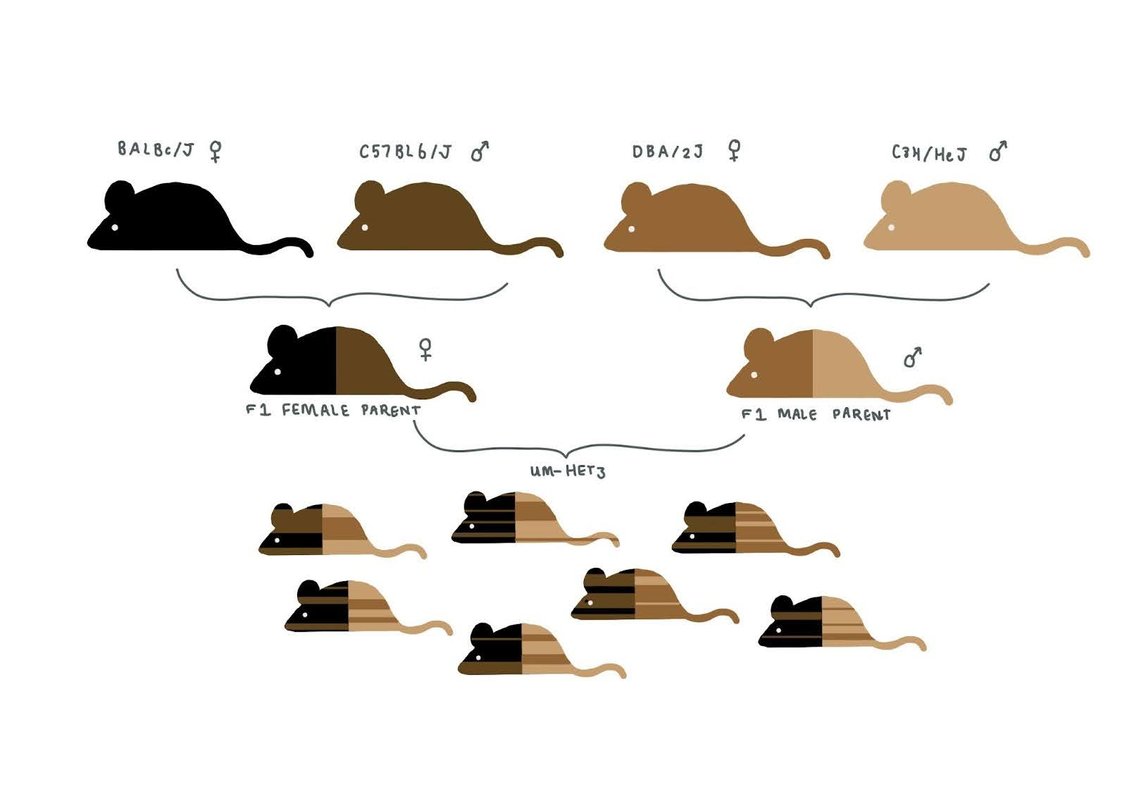
Another consideration in the choice of animal model was that it should demonstrate multiple causes of death. While outbred mice exhibit a greater range of pathologies at death than inbred mice, we noted that heterogenous mice are still imperfect models, dying of cancer more often than humans. One conjecture: perhaps mice’s longer telomeres, combined with their smaller overall cell count, enable more tumors to become lethal, even without telomerase activation?
We also discussed several study design and mouse husbandry choices. The untreated control group is larger than treatment groups because one control group is used across multiple treatments, increasing the statistical power achieved with the given budget. There are several trade offs associated with group or single-housing mice. It is easier to control dosage of food and reduce male fighting when you single-house mice, but in addition to being more expensive, isolation can cause mice to develop psychological issues. The ITP chose to group-house the mice but placed fewer mice in male cages (3 males) than in female cages (4 females) to reduce the impact of fighting.
The ITP data invites the question: why do most of the positive interventions have a greater effect in male mice? We don’t have a great explanation for this. It could be related to the existing survival advantage of females, which holds in HET-3 mice too: perhaps females are already more optimized to live longer than males? Another thing to consider in animal studies is the reproductive status of females, as this can affect their lifespans.
The new Rejuvenome project has a similar flavor to the ITP, but differs in several key ways. The ITP is limited to testing single compounds and focuses on lifespan as the primary endpoint (in Phase I). Conversely, the Rejuvenome plans to measure a large range of biomarkers per intervention, and is less restricted on the type of intervention (including compound interventions, and more expensive interventions out of scope of the ITP like injections). This is made possible by a larger budget per intervention. The Rejuvenome is also avoiding inbred mice, but using aged diversity outbred mice rather than HET-3 mice.
Mitochondria in aging (2004)

Paper(s): Nuanced review: Bratic, A., & Larsson, N. G. (2013). The role of mitochondria in aging. The Journal of clinical investigation, 123(3), 951–957.; Landmark paper for mutations: Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J. N., Rovio, A. T., Bruder, C. E., Bohlooly-Y, M., Gidlöf, S., Oldfors, A., Wibom, R., Törnell, J., Jacobs, H. T., & Larsson, N. G. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature, 429(6990), 417–423.
Motivation for including: Mitochondrial function is a major topic in aging biology, both because their metabolism produces substrates for other mechanisms (eg NAD+), as possible producer of reactive oxygen species (ROS), as a regulator of nutrient sensing, and more.
Journal Club Presenter: Lada Nuzhna
Slides: Link
Presentation date: 4/17/2022
How bad is the influence of mtDNA mutations?
It is pretty inconclusive:
mice without proofreading ability at mtDNA polymerase have significant accumulation of mutations and premature aging phenotype (which is probably a bad test for mtDNA mutations anyways, since the actual mutation load is very likely lower - see next bullet)
only mutation load of >60-80% leads to dysfunction but the actual mutation load in mtDNA rarely exceeds 1% (see papers “Mitochondrial threshold effects”, “Does premature aging of the mtDNA mutator mouse prove that mtDNA mutations are involved in natural aging?”)
mice with ≈4.7 Kbp mtDNA deletion uniformly distributed among various tissues:
accumulation of up to ≈60% mtDNA deletion in different tissues displayed no signs of mitochondrial dysfunction and disease manifestation
whereas tissues with >85% mtDNA deletion exhibited mitochondrial dysfunction and disease phenotypes (see papers “Generation of mice with mitochondrial dysfunction by introducing mouse mtDNA carrying a deletion into zygotes”, “Inter-mitochondrial complementation: Mitochondria-specific system preventing mice from expression of disease phenotypes by mutant mtDNA”)
“Mitochondrial point mutations do not limit the natural lifespan of mice” showed that mutation in mtDNA in wildtype occur 10x less often than was previously reported; - this study was for wildtype mice so might not be relevant to other strains/conditions.
Impact of mitochondrial dynamics on aging (fission and fusion)
Fission - mediated by Drp1 among other things
increased expression of fission proteins in mouse skeletal muscle has been linked to obesity (see “Mitochondrial fission contributes to mitochondrial dysfunction and insulin resistance in skeletal muscle”)
inhibition of Drp1 led to improved muscle insulin sensitivity as well as obesity protection in high-fat diets (see “Disruption of mitochondrial fission in the liver protects mice from diet-induced obesity and metabolic deterioration”)
Fusion - mediated by mitofusins Mfn1 and Mfn2 + inner membrane fusion proteins Opa1
Mfn1 is important for energy balance and thermogenesis
There is decline in Mfn2 expression in skeletal muscle as we age. This has been associated with age-related shifts in metabolism + onset of sarcopenia
Reduced expression of Mfn1 and Mfn2 has been linked to diabetes-2 and obesity
Removal of Mfn2 causes glucose intolerance and enhanced gluconeogenesis (liver-specific)
Things that go wrong with mitochondria as we age - final thoughts:
Somatic mtDNA mutations: can be bad if the mutation load is high, but it is not as high as we thought; some specific mutations are observed more often than others and are pretty bad.
Impaired oxidative phosphorylation: energy production is one of the primary functions of mitochondria and we get a gradual ATP decline as we age.
Oxidative damage: was big at the dawn of the aging field, but enthusiasm has been going down since then. People often mention oxidative damage when talking about mitochondria, but it is unclear whether ROS is higher in mitochondria than in any other part of the cell
Things to keep in mind: Assays to measure ROS are NOT robust, measuring the short lived ROS species itself is hard so ~all assays measure some downstream surrogate product.
Dysregulation of dynamics: From the studies linked above, it is pretty clear that dysregulated dynamics is detrimental for cells.
As a result of these things, we get decreased abundance and quality of mitochondria.
Heterochronic parabiosis (2005)

Paper(s): Primary paper: Conboy, I., Conboy, M., Wagers, A. et al. Rejuvenation of aged progenitor cells by exposure to a young systemic environment. Nature 433, 760–764 (2005); Historical perspective: Conboy, M. J., Conboy, I. M., & Rando, T. A. (2013). Heterochronic parabiosis: historical perspective and methodological considerations for studies of aging and longevity. Aging cell, 12(3), 525–530.
Motivation for including: Heterochronic parabiosis catalyzed the study of the role of circulating factors on age-related processes. It was one of the first interventions shown to have positive effects on aging phenotypes of old mice, with positive lifespan results as early as 1972.
Journal Club Presenter: Madison Ueland
Slides: Link
Presentation date: 4/03/2022
Summary
Parabiosis refers to the surgical conjoining of two animals (typically mice) allowing the development of a shared vascular system. When one animal is old and one is young, this is termed heterochronic parabiosis (HPB).
Several theories have arisen attempting to explain the benefit experienced by the older parabiont:
Dilution of deleterious factors in old blood (commonly termed the “dilution hypothesis”)
Addition of pro-youth factors present in young blood (occasionally called the “infusion hypothesis”
Benefit from young organs e.g. liver and kidney function
Different experiments can be used to distinguish between these theories:
Heterochronic blood exchange: blood exchange between mice without surgical attachment allows more controls (organs are no longer shared, timing is no longer dependent on development of vascular connections, precise amount of blood can be exchanged).
Neutral blood exchange / plasmapheresis: replacement of old plasma with artificial plasma solution composed of saline and albumin, enabling testing of dilution hypothesis without confounding by factors present in young blood.
Addition of putative pro-youth factors
Inhibition of putative pro-aging factors
Many putative factors have been investigated over the past decade, and some have led to the creation of companies e.g. the hypothesis that CCL11 is a pro-aging factor led to Alkahest. Nevertheless, there is little consensus, limited published human data and no clear positive results in humans.
GDF11 has garnered most attention, after initially being proposed as a pro-youth factor by Amy Wager’s lab at Harvard, results which were subsequently challenged by papers that questioned several claims, including whether circulating GDF11 levels even decrease with age. (These are detailed below.)
Background
Parabiosis was pioneered by Paul Bert in the mid 1800s. A century later, Clive McCay extended parabiosis experiments to aging by pairing old and young mice, termed heterochronic parabiosis (HPB). Despite positive survival results, where the older partners of the parabiotic pair outlived (female) or matched (male) both age-matched control parabionts and single controls, HPB fell out of favor for decades, in part due to the high mortality from ‘parabiotic disease’.
HPB was brought back into the fray in the early 2000s at Stanford and found to improve the regenerative capacity of liver, muscle and brain in the older parabiont mice (and detriment the younger parabiont). The aging field was nascent at the time: these papers pre-dated the positive lifespan results for late-life rapamycin from the NIA ITP, as well as senolytics and epigenetic clocks.
Despite much research in the last two decades, there is little consensus about preclinical results and no remarkable human data. The debate about GDF11 is a prime example: interest was spiked into GDF11 as a putative pro-youth factor by several high profile studies purporting that GDF11 levels declined with age and restoring them improved aging phenotypes such as the activity of satellite cells (muscle stem cells). Soon after, other studies questioned these results, proposing that circulating GDF11 levels actually increase with age, and that the original assay to quantify GDF11 was non-specific. They also found that GDF11 inhibits satellite cell proliferation (consistent with the well-understood actions of a highly homologous ligand, myostatin). A window into the debate is captured below:
| Paper | Affiliation | Direct Observations | Interpretations |
|---|---|---|---|
| Loffredo et al., 2013 | Harvard (Wagers, Lee) |
* n=3 in each group for Western blot (Fig 7A) ** used nonspecific antibody |
|
| Sinha et al., 2014 | Harvard (Wagers, Lee) |
|
|
| Egerman et al., 2015 | Novartis |
*Differences in source and quality of recombinant GDF may explain why Sinha et al. (2014) results were inconsistent |
|
| Hinken et al., 2016 | GSK, Five Prime Therapeutics |
|
Unable to reproduce Sinha et al., consistent with the small decrease in number of cells found in Egerman et al following recombinant GDF11 treatment. |
| Rodriguez et al., 2016 | Universidad de Oviedo (Spain) |
* small sample size (5 males, 8 females treated with rGDF11; 6 males and 6 females vehicle-treated). |
|
Discussion Summary
The two primary theories explaining the positive effects of parabiosis on old mice - the dilution and infusion hypotheses - are not mutually exclusive. However, most researchers reside in one camp or another. As always, it is important to be wary of research origins and conflicts of interest, especially when commercial interests are involved. The GDF11 case study is reminiscent of the sirtuin controversy, where the bulk of positive data originated from a single lab.
Although human data is typically preferable to preclinical studies, it is important to keep in mind several downsides. Firstly, sample size is often small, as it is with Therapeutic Plasma Exchange (n=9 in PLASMA study). We should avoid being swayed by underpowered human data, and to do so it may be necessary to consciously discount underpowered studies given the probability of bias for publishing/announcing seemingly positive results. Secondly, although results of clinical trials are meant to be published on clinicaltrials.gov, this is rarely enforced. If studies have concluded and results have not been published - even if there is a vaguely upbeat press release on the sponsor’s website - it is likely that the results were negative.
One possible complication for the field of parabiosis is that strong rejuvenation could be the result of multiple pro-youth factors, with minor/no effects individually. This would greatly complicate reductionist experiments looking for such factors.
Nutrient Sensing

Paper(s): Finkel, T. The metabolic regulation of aging. Nat Med 21, 1416–1423 (2015).
Motivation for including: The strongest intervention effects on lifespan (including insulin signaling, mTOR, and dietary restriction) all relate to nutrient availability. It’s important to understand how central this aspect of biology is to aging research, even if we don’t truly know why. This journal club reviews the primary pathways linking nutrients to longevity, aiming to contextualize previous and future journal clubs diving into primary data on metabolic interventions, such as Cynthia Keynon’s daf-2 mutant C. elegans, the sirtuins/resveratrol controversy, rapamycin and CR in humans.
Journal Club Presenter: Madison Ueland
Slides: Link
Presentation date: 2/06/2022
Takeaways
Insulin/IGF-1 signaling (ISS) was the first pathway linked to lifespan with Tom Johnson’s age-1 (homologous to mammalian PI3K) mutants and Cynthia Kenyon’s daf-2 (homologous to IGF-1 receptor) mutants.
Inhibiting IIS lengthens lifespans in worms and flies.
In mice, targeted approaches altering IGF-1 receptor or IGF-1 bioavailability increase longevity in some strains, with sex-specific effects. Full-body knockouts of IIS genes are often lethal, tissue-specific modulation more promising.
Correlative evidence in humans: mutations in IGF-1 receptor associated with exceptional longevity, and low IGF-1 levels in female nonagenarians associated with longer survival.
Mammalian Target of Rapamycin (mTOR)
Inhibiting mTOR extends lifespan in yeast, worms, flies and mice, but not without side effects (can impair healing, cause cataracts and testicular degeneration in mouse models)
In yeast, worms and flies, adding dietary restriction to inhibition of TORC1 (of the two TOR protein complexes, TORC1 is most studied in aging) does not further increase lifespan: mTOR mediates CR benefits?
NAD+ and sirtuins
NAD+ is a central cofactor in metabolism, and declines with age.
In the early 2000s, there was much attention surrounding sirtuins, NAD+ dependent deacetylases of histones and non-histone proteins. See sirtuins / resveratrol JC.
Supplementation with NAD+ precursor (NAM) did not extend lifespan of male mice.
AMPK is a sensor of nutrient-restricted states and catabolism.
Metformin activates AMPK in mice and worms.
In worms and flies, some kinds of dietary restriction require activation of AMPK orthologs to achieve lifespan extension.
Upregulating AMPK induces autophagy, a process that is also implicated in interventions altering other pathways.
Many longevity pathways are closely connected or cross-regulated (see diagram), making nutrient sensing networks difficult to untangle.
Important to be precise: effects often differ between tissues, species, sexes, and doses.
Discussion Summary
Discussed biases that plague many reviews, from optimism to selective reporting. This can happen innocuously because positive results are more likely to be well-known, or less innocuously when researchers use reviews to promote the importance of their field.
How can we best represent this information?
Graph-based diagrams are helpful, but it is challenging to include sufficient nuance and caveats.
An ideal diagram would not just include positive results, but also distinguish between relationships that have not been tested and those where no effect was found, in each case weighting the arrow by the strength of evidence.
Specifying the animal model and relevant tissue is important.
Given the nonlinearity and pleiotropy underlying nutrient signaling pathways, how optimistic should we be about metabolic interventions for longevity?
There are still grounds to be optimistic despite the messiness because evolution has already evolved life extension in response to caloric restriction. We’ve seen it work.

Nutrient Sensing Pathway Diagram (source)
Late-life rapamycin intervention (2009)

Paper(s): Harrison, D., Strong, R., Sharp, Z. et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460, 392–395 (2009).
Motivation for including: Two-fold: Rapamycin has robustly extended lifespan of multiple species by inhibiting mTOR, an apparent node of aging. Also, this paper added strong data for a common doubt about whether aging could be ‘reversed’ or only slowed down through lower metabolism/damage accumulation.
Journal Club Presenter: Kush Sharma
Slides: Link
Presentation date: 2/06/2022
Summary
Rapamycin is an mTOR inhibitor
mTOR is a central metabolic regulator that turns the cell towards either growth/proliferation or resource conservation depending on environmental and internal conditions
Rapamycin had been shown to extend lifespan in many non-mammalian model organisms
This study was the first to rigorously examine the effects of mTOR on mammalian lifespan via the NIA ITP (see previous presentation for details), in genetically heterogenous mice
It found unambiguous lifespan extension in mice, with sex-dependent effects
Importantly, rapamycin treatment was begun late in life, and was the first solid example of large late-life effects, meaning that we can still delay aging when begun late in life
It left unclear the impact of dosing, dosing schedule, and initiation time on lifespan since only one regimen was tested
Background / Context
Rapamycin is a pharmaceutical compound that inhibits the mTOR pathway. The mTOR pathway is a central metabolic regulator: it senses nutrient and energy levels, and if there are sufficient nutrients, directs the cell to grow, and otherwise conserves resources, e.g. via autophagy. There are two mTOR complexes: mTORc1 and mTORc2. It’s suspected that mTOR inhibition is one of the central mechanisms of caloric restriction’s lifespan-extending effects. mTOR inhibition via rapamycin, prior to this study, had been shown to extend lifespan in a variety of model organisms but had not been tested in mammals. Additionally, much of the aging field implicitly or explicitly operated with something like a wear and tear model where aging could only be slowed, and late-life intervention would be difficult or impossible. The importance of this paper was in showing that lifespan extension late in life was possible in mammals.
mTOR Pathway in Figures
All figures are from mTOR at the nexus of nutrition, growth, ageing and disease, which is an excellent review for more detail on the mTOR pathway.
1. Function of mTORc1 and mTORc1

2. Downstream targets of mTORC1 and mTORC2 signaling


3. Upstream regulators of mTOR

Caloric restriction in primates extends/doesn’t extend lifespan (2009-2012)

Paper(s): Colman, R. J., Anderson, R. M., Johnson, S. C., Kastman, E. K., Kosmatka, K. J., Beasley, T. M., Allison, D. B., Cruzen, C., Simmons, H. A., Kemnitz, J. W., & Weindruch, R. (2009). Caloric restriction delays disease onset and mortality in rhesus monkeys. Science (New York, N.Y.), 325(5937), 201–204. & Mattison, J., Roth, G., Beasley, T. et al. Impact of caloric restriction on health and survival in rhesus monkeys from the NIA study. Nature 489, 318–321 (2012).
See also joint summary Mattison, J. A., Colman, R. J., Beasley, T. M., Allison, D. B., Kemnitz, J. W., Roth, G. S., Ingram, D. K., Weindruch, R., de Cabo, R., & Anderson, R. M. (2017). Caloric restriction improves health and survival of rhesus monkeys. Nature communications, 8, 14063.
Motivation for including: Dietary restriction (of different types, including calorie restriction, protein restriction, and intermittent fasting) has shown lifespan extension in many organisms and is considered by some to be the #1 longevity intervention. These studies are the most relevant data we have for possible human translation.
Journal Club Presenter: Tara Mei
Slides: Link
Presentation date: 2/20/2022
Summary
1. CALORIC RESTRICTION (CR) EXTENDS LIFESPAN (Colman et al. 2009)
Methods
1989, Wisconsin National Primate Research Center, University of Wisconsin Madison (UWisc).
76 monkeys. 46 males, 30 female adults (7-14 years old at start, maximum lifespan 40 years).
Monkeys randomised to control (38 monkeys) or CR (38 monkeys) diets (10% baseline food intake reduction per month over a three-month period to achieve a 30% total restriction) based on baseline food intake, weight, age.
Dead animals underwent a necropsy by a pathologist blinded to diet group to determine cause of death (Note: it is likely that pathologists would nonetheless be aware of diet group based on adiposity of the dead animals).
Results
Aging pathologies
Reduced sarcopenia, diabetes [no glucose homeostasis impairment in CR monkeys vs ~16/38 in control monkeys], neoplasia/cancer growth [50%], cardiovascular disease [50%], brain atrophy [in regions that subserve motor function and assets of executive function]
Control monkeys experienced aging diseases at 3x the rate of CR monkeys
Death
37% of control animals vs 13% of CR animals died of age-related causes over the 20-year study period. Others (7 control animals, 9 CR animals) died of non-age-related causes, e.g. anaesthesia complications, gastric bloat, endometriosis, injury.]
[CR affects overall mortality in predicted direction, but not in a statistically significant way]
[The most prevalent aging diseases for these monkeys are diabetes, cancer, and cardiovascular diseases, similar to those in humans]
2. CALORIC RESTRICTION DOESN’T EXTEND LIFESPAN (Mattison et al. 2012)
Methods
1987, National Institutes of Health (NIH) Animal Center.
121 monkeys in 3 age categories (juvenile, adolescent/adult, old).
Monkeys randomised to control or CR diets (30% total restriction compared to sex-, age-, and initial body size-matched controls) based on baseline food intake, weight, age.
Dead animals underwent a necropsy by a pathologist to determine cause of death.
Results
Aging pathologies
Control and CR monkeys showed no difference in causes of death: Neoplasia, cardiovascular disease, amyloidosis, general organism deterioration
CR benefits: lower weight, triglycerides, cholesterol, fasting glucose, oxidative stress (measured by plasma free isoprostane)
Death
Old-onset CR monkeys did not live longer than controls [both sexes]. CR negatively affected young-onset CR monkeys, who showed delayed maturation and slowed skeletal growth.
Why the difference?
All NIA monkeys consumed fewer calories than UWisc control monkeys. Body weight was lower in NIA control monkeys (about equivalent to UWisc CR monkeys) and CR monkeys than in UWisc control monkeys.
Study design differences:
Different diet composition. NIA: Natural ingredient base, WNPRC: Purified diet (each ingredient supplies a specific nutrient; each required mineral and vitamin added as a separate component).
Different nutrient sources. Protein—NIA: wheat, corn, soybean, fish, alfalfa meal; WNPRC: lactalbumin. Fat—NIA: soy oil, oils from other natural ingredients like corn, wheat, fish; WNPRC—corn oil. Carbohydrate—NIA: ground wheat, corn; WNPRC: corn starch, sucrose. NIA diet also contained flavonoids.
NIA CR and CON monkeys received vitamin- and mineral-supplemented diets; only WNPRC CR monkeys received vitamin- and mineral-supplemented diets
NIA CON monkeys were not truly fed AL. The regulated portioning for CON monkeys may represent a slight CR.
CR-induced lifespan extension may be a laboratory phenomenon: CONs are overfed, and CR just optimises survival to what we would see in non-laboratory conditions.
NIA monkeys were more genetically diverse (China and India) than WNPRC monkeys (India).
Different feeding times
UWisc treated monkeys that developed chronic disease, whereas NIA did not unless monkeys suffered from acute pain and endometriosis. It is thus plausible that CR delays the onset of age-related diseases, but decreases robustness to them.
Discussion summary
UWisc tried to disentangle age-related mortality from other types of mortality, though aging increases the likelihood that you’ll die from any other mortality cause (e.g. injury).
NIA: Animals that died of “non-age-related causes”, e.g. death from anesthesia, were censored from mortality and morbidity analyses. But does CR affect these “non-age-related” things? Unclear, but one study does show an effect of short-term intermittent fasting on the duration of anaesthesia in mice (Shahroozian et al. 2017)
Though UWisc monkeys were fed AL and NIA monkeys were not, calorie consumption actually seems quite similar between the two.
Relevant variables not considered: timing/duration of CR (e.g. CR 1x/week vs constant restriction), type of nutrient restricted e.g. protein, impact of quality of initial diet on value of CR
Are demographics that eat a lower-calorie diet than the typical Western diet (e.g. Japanese) considered calorically restricted? Possibly, but hard to disentangle from other sociodemographic factors and genetics.
Clearance of senescent cells improves health (2011)
Paper: Baker, D., Wijshake, T., Tchkonia, T. et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236 (2011). This was the paper that launched Unity Biotechnology. Later shown in WT mice Baker, D., Childs, B., Durik, M. et al. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 530, 184–189 (2016).
Motivation for including: Senescent cells are a major area of research and therapeutic development in aging biology. The idea that a subset of ‘bad cells’ drive (parts of) aging is appealing, since these cells could be targeted, and there’s strong data. However, the term ‘senescence’ has some ambiguity.
Journal Club Presenter: Tara Mei
Slides: Link
Presentation date: 3/27/2022
Summary
Question: What happens if you remove senescent cells (SCs)?
Baker et al. 2011
Methods:
1. Made a transgene that removes p16Ink4a-positive SCs in progeroid (BubR1H/H: shortened lifespan, exhibit age-related phenotypes, such as infertility, lordokyphosis, sarcopenia, cataracts, fat loss, cardiac arrhythmias, arterial wall stiffening, impaired wound healing and dermal thinning) mice upon drug treatment.
2. Administered drug that clears SCs (at a. weaning age, b. 5 months).
3. Examined effects of clearing SCs on aging.
Results:
a. SCs cleared at weaning age
Delayed lordokyphosis and cataracts, larger muscle fibers, larger fat deposits, better exercise (duration, distance traveled, overall amount of work performed). No overt side effects. No marked increase in overall survival. Authors suggest this is because cardiac failure is the main cause of death in the studied mice, and contributors to cardiac failure such as cardiac arrhythmias and arterial wall stiffening are p16-independent—as such, SC clearance has no effect on those contributors and thus no effect on lifespan.
b. SCs cleared at 5 months
Unchanged cataracts, larger muscle fibers, larger fat deposits, better exercise
Baker et al. 2016
Methods:
Similar to previous study, except SCs cleared at 1 year in wildtype rather than progeroid mice.
Results:
Longer lifespan in both sexes (lifespan increase from ~just under 2 years vs just over 2 years), delayed cataracts, larger fat deposits, reduced glomerulosclerosis, increased cardiac stress tolerance.
Discussion:
Given the heterogeneity of SCs and the fact that there is no unique biomarker for SCs, how do we “know” that we are even clearing “SCs”?
Is senescence always “bad”? See Grosse et al. 2020: elimination of SCs caused liver and perivascular tissue fibrosis.
It is important to note what a paper does and doesn’t show—look at the data, not just the conclusion implied in the abstract. For example, Baker et al. 2016 doesn’t show SC clearance in the liver or the colon, so any lifespan extension resulting from SC clearance probably isn’t from SC clearance in the liver or colon.
A few studies in humans
Lee et al. 2021: COVID-19 patients display SC biomarkers and elevated SASP levels.
Nehme et al. 2020: SCs may increase COVID-19 severity and decrease vaccine efficiency
Iske et al. 2020: Senolytics repair age-related damage in organs following transplantation
Justice et al. 2019: Senolytics improve physical function in idiopathic pulmonary fibrosis patients (preliminary study, N=14)
Hickson et al. 2019: Senolytics cleared SCs in kidney disease patients (preliminary study, N=9)
The (over)influential Hallmarks of Aging framework (2013)

Paper(s): López-Otín, C., Blasco, M. A., Partridge, L., Serrano, M., & Kroemer, G. (2013). The hallmarks of aging. Cell, 153(6), 1194–1217.
Motivation for including: This 2013 review paper is the first stop for many newcomers to the field, and the mechanism-centric paradigm it proposes permeates everything from conference sessions to startup companies. However, the mechanisms do not serve as a list of necessary and conditions for aging, so the paradigm isn’t a complete explanation.
Journal Club Presenter: Divya Cohen
Slides: Link
Presentation date: 4/30/2022
Summary
Goal for Hallmarks paper
Understand the causal factors for the time-dependent accumulation of cellular damage of aging
Criteria for inclusion
Manifest during normal aging
Experimental aggravation should accelerate aging
Experimental amelioration should retard aging
Ideally in multiple models including mice and humans
Summary of 9 Hallmarks of aging
Hierarchical (according to authors)
Primary (causes of damage): genetic instability, telomere attrition, epigenetic alterations, loss of proteostasis
Antagonistic (response to damage): deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence
Integrative (culprits of the phenotype): stem cell exhaustion, altered intracellular communication
Background / Context
In 2000 a paper was published with 6 hallmarks (later 10) for Cancer that were very clarifying for the field and directed future therapeutic development. The hope was the Hallmarks of Aging would do the same for the aging field. One difference is that the Hallmarks of Cancer are necessary and sufficient features for a cell to be cancer, whereas the hallmarks of aging are important mechanisms with different degrees of specificity and with interactions and potentially hierarchies between them. The evidence provided for them is largely correlative. Future studies need to show loss or gain of function in animal models to move beyond correlative analysis and provide causal evidence. So although helpful, they have been less clarifying than the cancer hallmarks that were useful for therapeutic development.
This is part of the reason the Longevity Curriculum as described in this Longevity Journal Club Book does not seek to recapitulate each Hallmark of Aging.
Discussion Summary
Below is a table that provides the level of evidence that existed in 2013 compared to 2022 as per the criteria developed in the study (see Summary above):
| Hallmark of Aging | Evidence Level in 2013 | Evidence Level in 2022 | Notes from Paper |
|---|---|---|---|
| Genomic Stability | a and b |
a and b (still no c with 50 years of experiments) |
|
| Telomere Attrition | a, b and c but c is only available in mice | a, b and c but c is only available in mice |
|
| Epigenetic Alterations | a, b and c but c is only available in mice | a, b and c but c is only available in mice (unsure role of methylation clocks, some epigenetic modifiers do not change lifespan so how important is e.g. SIRT6 and as a corollary this hallmark of aging) |
|
| Loss of Proteostasis | a, b and c but c is only available in mice | a, b and c but c is only available in mice (but more focus recently on autophagy, other study on chaperones in mice but not reproducible) |
|
| Deregulated Nutrient Sensing | a, b and c but c is only available in mice | a, b and c but c is only available in mice (but interventions in mice highly dependent on food, type of mice, gender.) |
|
| Mitochondrial Dysfunction | a and b | a and b |
|
| Cellular Senescence | a and b (authors propose a & b, and c but only mice) | a, b and c but c is only available in mice |
|
| Stem Cell Exhaustion | a and c | a and c (maybe only important in highly proliferative tissues) |
|
| Altered Intercellular Communication | a, some b and c | a, some b and c |
|
Final remarks
Hallmarks of Aging are largely correlative
Loss or gain of function in animal models needs to move beyond correlative analysis and provide causal evidence, within and between hallmarks.
Hallmarks are highly interlinked
Possibly should consider new Hallmarks like extracellular crosslinks i.e. glycans. (Multiple papers have proposed extra hallmarks).
We don’t know yet if these are necessary and sufficient conditions
For reference, other papers that describe frameworks of aging:
Horvath methylation clock (2013)

Paper(s): Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol 14, 3156 (2013).
Comments: Methylation clocks are the most popular approach (currently) to measuring ‘age’ from both cellular and tissue samples. If they work as intended, they would provide both a way to measure cellular age in high-throughput (e.g. for screening), and potentially a noninvasive biomarker for trial. Big promise!
Journal Club Presenter: Lada Nuzhna
Slides: Link
Presentation date: 2/03/2022
Context
We have methylation of histones (proteins around which DNA is wrapped) and methylation of DNA itself. For ‘methylation clocks’, DNA methylation is used.
While methylation was studied since 70s, Horvath’s paper in 2013 was the first one to show its potential connection to aging. Since 2013, we have seen many new clocks. They broadly can be summarized into first-generation (cross-sectional data: take people of different ages and see which CpG sites predict age) and second-generation (longitudinal data: take people and measure their methylation over time to see which sites predict age).
Overall, if you trust metrics used to assess the clocks, we already have clocks that are good enough. At the same time, for the clocks used to predict chronological age, perfect metrics do not matter as much - after all, that is already a known metric. Publication of new clocks slowed down during the past year or two and current research is mostly oriented towards understanding what the clocks mean, how they work over time, and in different diseases.
People
The researchers that have done the most work on clocks are:
Steve Horvath (author of the first methylation clock paper)
Morgan Levine (she was part of Horvath’s lab but later started working on it independently)
Vadim Gladyshev (scAge)
Why were clocks created around DNA methylation and not any other epigenetic modification (or even transcriptome) ?
DNA methylation (DNAm) is unevenly distributed and, in contrast to many other epigenetic marks, is heritable and more stable. Its role as a gene suppressor was first discovered by correlating gene expression and DNAm (see DNA methylation sites in the vicinity of the chicken β-globin genes (1979); Patterns of integration of viral DNA sequences in the genomes of adenovirus type 12-transformed hamster cells (1978); De novo methylation and expression of retroviral genomes during mouse embryogenesis (1982) ). Correlation experiments weren’t sufficient to infer the functional role though - it was only after experimental inhibition or deletion of DNA methyltransferases, in vitro methylation of DNA and genetic deletion of differentially methylated regions that its role was confirmed. All of this was possible because we already had assays for profiling methylation.
There was also an effort to create a transcriptome clock. However, the transcriptome is not fully heritable and is dynamic. Thus, it would be harder to differentiate between stable long-term changes and dynamic short-term reactions to the environme
How does methylation influence aging?
The causal role of methylation in aging is not confirmed yet (we are funding this in round 2 of Impetus Grants) and, while some companies have been already using clocks in clinical trials (the one I came across was iAge), clocks aren’t widely used as biomarkers yet (you can read more on this in “What do methylation clocks need to become biomarkers in aging trials?”). The overall pattern is:
global DNAm declines with age -> loss of transcriptional control, deleterious aging effects;
DNAme often increases (=hypermethylation) at CpG islands.
Keep in mind that most datasets were constructed for bulk measurements and are susceptible to different statistical paradoxes (like a Simpson paradox). Single-cell measurements are more powerful, but are also more sparse, which makes it hard to construct a clock from them (although there was a recent effort to overcome this sparsity - see scAge from Gladyshev’s lab).
So what do the clocks tell us about aging?
The research on phenotypic interpretation of CpG sites that are picked up by clocks is lagging. Making such studies might be complicated given that:
different clocks point to different CpG sites as most meaningful for predicting age or aging (see “Many chronological aging clocks can be found throughout the epigenome: Implications for quantifying biological aging”).
genes are regulated by many other things (for example topologically, by interacting with enhancer regions), so we need to have tools that measure multiple epigenetic influences at the same time to figure out whether it's just methylation affecting a particular gene.
What is immune aging? (2013-)

Book Chapter: Müller, L., Di Benedetto, S., Pawelec, G. (2019). The Immune System and Its Dysregulation with Aging. In: Harris, J., Korolchuk, V. (eds) Biochemistry and Cell Biology of Ageing: Part II Clinical Science. Subcellular Biochemistry, vol 91. Springer, Singapore.
Motivation for including: The term immune aging is used, somewhat ambiguously, to cover multiple processes that tie the immune system to aging: loss of the thymus, loss of stem cell diversity, loss of T cell function, as well as reduced functional capacity of the immune system. We want to delineate and clarify these.
Journal Club Presenter: Kush Sharma
Slides: Link
Presentation date: 5/8/2022
The discussion for this session was minimal and touched on the following points.
Should we be using mice to study immune aging?
There are two points here. First, lab-raised mice are quite different from mice raised in the wilderness. Lab-raised mice have not been exposed to the range of pathogens that wild mice, and humans, have been exposed to in their natural environments. As a result, adult lab-raised mice have immune systems closer to infant humans, rather than adult humans. E.g. lab-raised mice have very few memory CD8+ T-cells, unlike wild-raised mice. This influences the applicability of studies done on lab-raised mice to humans.
Is decreased clearance of senescent cells also a consequence of immune aging?
Immune cell responses are modulated by senescent cells via the SASP. There’s some evidence that impaired immune surveillance as a result of immune aging causes an accumulation of senescent cells.
How many hallmarks of aging do you imagine being downstream of immune function?
This is a difficult question to untangle because there are immune cells in every tissue, and if the immune cells are in a chronic state of activation and perturbing the environments they’re in, they’re leading to some of the damage that we call aging. In the literature, immune cell activation can be linked to all of the hallmarks of aging.
Partial rejuvenation in vivo helps progeria mice (2016)

Paper(s): Ocampo, A., Reddy, P., Martinez-Redondo, P., Platero-Luengo, A., Hatanaka, F., Hishida, T., Li, M., Lam, D., Kurita, M., Beyret, E., Araoka, T., Vazquez-Ferrer, E., Donoso, D., Roman, J. L., Xu, J., Rodriguez Esteban, C., Nuñez, G., Nuñez Delicado, E., Campistol, J. M., Guillen, I., … Izpisua Belmonte, J. C. (2016). In Vivo Amelioration of Age-Associated Hallmarks by Partial Reprogramming. Cell, 167(7), 1719–1733.e12.
Motivation for including: The promise of resetting multiple aspects of cellular aging by partially activating the Yamanaka (or other transcription) factors has recently received a lot of attention and investment.
Journal Club Presenter: Divya Cohen
Slides: Link
Presentation date: 3/13/2022
Summary
Yamanaka and Kazutoshi Takahashi in 2006 sought to find the minimum set of reprogramming factors needed to produce pluripotency in somatic cells
Identified 24 transcription factors known to cause pluripotency
Tested each one separately, but no drug-resistant colonies found
Tested all 24 together, found not obtain any drug-resistant colonies
Then took one away at a time until got minimum required:
Oct4, Sox2, Klf4, and c-Myc (OSKM)
Juan Carlos group in 2016 sought to rejuvenate progeria mice in vivo safely
They first created progeria model organism in the form of LAKI 4F mice
Used premature aging mouse model with G609G mutation of the gene Lmna knocked-in (LAKI)
Causes accumulation of a truncated form of lamin A (called progerin) which is responsible for human HGPS
To enable inducible expression of the Yamanaka factors upon doxycycline treatment, LAKI mice were crossed to mice carrying an OSKM polycistronic cassette (4F) and a rtTA trans-activator thereby generating LAKI 4F mice.
Found partial reprogramming cyclically can be done safely without creating teratomas in mice
Showed cyclic OSKM induction protocol, consisting of 2 days of doxycycline administration followed by 5 days of doxycycline withdrawal had lower mortality even after 35 cycles
This can rejuvenate progeria mice i.e. lifespan extension
Protocol included cyclic OSKM induction in LAKI 4F mice beginning at 8 weeks of age for 6 weeks hence measured result at 14 weeks
Resulted in increase in median and maximal lifespan, reduction in spine curvature and improvements in skin, liver, stomach, and kidney
It has shown signs of rejuvenating some aspects of wild type mice but the case is not as strong
Method included three cohorts of mice (and controls):
long term - 12 months with 10 mo Rx, OR 7 months with 15 mo Rx
short term - 25 month with 1 month Rx
Results
Methylation clocks revealed a significant reduction of epigenetic age in skin (but not other tissues)
Follow up with Excisional Wound Splint in the skin showed No Difference in closure rate of the wound
Newer papers by Sinclair lab 2020 seems to have attempted systemic OSK delivery but did not report lifespan effects and Ocampo 2022 (pre-print) showed OSKM continuous delivery causes specific harm to liver and intestines leading to mortality.
Background / Context
Dr Yamanaka started the field of reprogramming in 2006 with the publication of a landmark paper which for the first time described four transcription factors (Oct4, Sox2, Klf4, and c-Myc (OSKM)) that are necessary and sufficient for producing pluripotency in somatic cells. He first proved this in mice in 2006 and later in human cell lines in 2007, and won the Nobel Prize for his work in 2012. Juan Carlos' group later expanded this work with partial reprogramming in 2016 and showed it could be used for rejuvenation in vivo in progeria mice safely without creating teratomas but notably the work did not include lifespan extension.
Discussion Summary
GSEA (Gene Set Enrichment Analysis) is a “computational method that determines whether an a priori defined set of genes shows statistically significant, concordant differences between two biological states (e.g. phenotypes)”. Link
It was the default analysis of every -omic here.
It is notable that all the analysis in this paper is using bulk sequencing and -omics. So it is not possible to tell whether any change in gene expression is from individual cells up or downregulating versus different types of cells being present leading to genes "upregulated" if they're uniquely found in certain cell types.
It is also possible since in vitro reprogramming kills some cells that in vivo reprogramming could also just be a type of selection. We still do not know.
It could be that reprogramming effectiveness is not the same in all tissues. As evidenced by the recent Ocampo 2022 (pre-print) reprogramming can be much more lethal with expressions in specific organs such as the liver and intestines.
These experiments lead to more questions on if reprogramming is helpful in normal aging processes. Next steps to elucidate this further could be manipulating doses but this is a large logistical challenge.
Could try to speed up discovery either with selecting transcription factors better like e.g. GC Therapeutics, Bit.bio, Mogrify or high-throughput testing like Gordian.
Human GWAS studies (2019)

Paper(s): Overview: Melzer, D., Pilling, L.C. & Ferrucci, L. The genetics of human ageing. Nat Rev Genet 21, 88–101 (2020).
Motivation for including: Model organism work has shown us that genetics can modulate lifespan. How about human genetics? Only ApoE and FoxO3 have been consistently linked to long lifespan in large studies, but the review covers narrower findings.
Journal Club Presenter: Divya Cohen
Slides: Link
Presentation date: 2/27/2022
Background / Context
This paper is a review of GWAS studies in order to understand the heritability of aging. GWAS is a study of participants to identify statistical associations between genetic variants and traits of interest. It reaffirmed the nature vs nurture debate, in that aging too is a mixture of both and that the wide range of human lifespan cannot be just heritable or genetic.
Overview
Human aging is driven by a balance of damage and repair processes, influenced by both environmental exposures and genetic variation between people
Heritability reported ranges from Centenarians (48% in men and 33% in women), Twin Studies (20-30%), larger studies (16%), removal of assortative mating (10%)
Twin studies of common age- related diseases also have substantial heritable components: hip osteoarthritis ( 68%), T2DM heritability (61–78%), Alzheimer disease (58–79%), cardiovascular disease heritability (45–69%)
GWAS studies of common age- related diseases also have substantial heritable components: hip osteoarthritis (51%), T2DM heritability (18%), Alzheimer disease (7.1%).
Missing Heritability Gap (Twin study - larger studies) - lack of coverage of several types of genetic variation including rare large- effect variants in GWAS, but also to possible overestimates in pedigree- based analyses
None of the variants identified thus far as being associated with (extreme) longevity seem essential (that is, not all long- lived people harbor them) and none seem sufficient to achieve longevity (as all are fairly common in groups who die earlier). This finding is consistent with the notion that the heritable component in the longest 10% for survival is a quantitative trait likely affected by large numbers of small effect variants.
A whole genome risk score from the 1 million lifespan parental longevity GWAS, comparing individuals in the top and bottom deciles, was associated with an increase of 3–5 years in life expectancy.
Discussion Summary
The earlier part of the discussion focused on understanding various definitions:
What is heritability? Correlation between your outcome and your parent's outcome.
What is the "Missing heritability gap": You can calculate heritability from the phenotype (lifespan, or whatever), controlling for socio demographics as best possible. You can also look for individual variants with odds ratios, and add those up. The heritability gap is the difference.
Note this could be for multiple reasons: Haven't detected all the important genes, e.g. because some are below (arbitrary) significance threshold, but also because we're basically testing for linear effects but effects could be non-linear or combinatorial. Not aware of tools to test. And equally could be because your phenotype estimate is wrong because something you didn't control for.
GWAS studies are very heavy on statistics. The actual test used in this paper is unknown but presumed to be common and hence it wasn't discussed. There was a discussion on how useful it is to go deep on statistics to be an aging biologist. Please note the Statistics of Biology section.
There was a discussion for ApoE4 and FoxO3. ApoE4 is the only one found in the exonic region and with the greatest effect, however, still not very large. Unsure why the article did not discuss FOXO3 in more detail. Does ApoE4 affect lifespan when you control for Alzheimers? Yes.
Measuring aging in humans (2019)

Paper(s): Ferrucci, L., Gonzalez-Freire, M., Fabbri, E., Simonsick, E., Tanaka, T., Moore, Z., Salimi, S., Sierra, F., de Cabo, R, Measuring biological aging in humans: A quest. Aging Cell 19:e13080 (2020).
Journal Club Presenter: Tara Mei
Motivation for including: Probably the biggest reason we don’t have more clinical trials for aging in humans is that we don’t have a (proven) direct measure of “aging”, which makes for unwieldy trials that must use indirect measures (e.g. incidence of multiple diseases) and/or seek to validate readouts as part of the trial. This paper presents progress and possibilities.
Slides: Link
Presentation date: 9/11/2022
Summary
“Developing an index of biological aging is perhaps the most critical milestone required to advance the field of aging research.” Some reasons:
1. Test the geroscience hypothesis that aging causes disease. An aging biomarker will enable us to test if individuals that accumulate diseases faster than does the general population show a faster change in this aging biomarker, i.e. age faster.
2. Enable preventative care and refine therapeutic choices. Current medical care focuses on diagnosing and managing diseases that are already symptomatic, so they are addressing symptoms rather than the underlying cause. Patients often come to the clinic when they are already suffering from many diseases and have lost their independence. Measuring the underlying causes of aging and thus disease will help doctors prescribe therapeutic choices more accurately and earlier. Such an understanding will also fortify the knowledge of geriatricians who face the challenge of applying the status quo “one-by-one” norm of clinical guidelines to patients who present long lists of diagnoses and impairments that very likely reflect the occurrence of more than one disease.
3. Identify genetic, environmental, and behavioral risk factors associated with aging. We could relate changes to an aging biomarker with triggers such as pollution exposure.
Currently available aging biology measures (based on Hallmarks)
1. Genomic instability [Thesis: Somatic mutations accumulate with age, affecting protein fidelity and the regulation of gene expression]
Evidence for link to aging:
Lacking, but there is strong evidence showing that somatic mutations accumulate with age (e.g. in B lymphocytes, skeletal muscle satellite cells, neurons; functional relevance of mutations unknown).
Available measures:
Quantification of DNA repair capacity is as yet unsatisfactory. Assays have not been independently verified nor applied to large populations. Most assays only address DNA repair capacity of a subset of specific lesions. There is no consensus on gold standard assays.
2. Telomere length [Thesis: Telomeres shorten during each cell division → bad things]
Evidence for link to aging:
Lacking. A 13-year study reported that average telomere length does shorten with age, but the direction and magnitude of change is cell-dependent and very heterogenous across individuals. There is also no evidence that telomerase, which replenishes lost telomeric DNA, is a resilience mechanism for aging. But telomere shortening does seem to be linked to a number of bad outcomes, like increased CVD, reduced immune response to influenza vaccination, and obesity.
Available measures:
There exist several methods to measure telomere length, e.g. restriction fragment analysis and fluorescence in situ hybridisation. But observational studies have reported contrasting results and erratic changes over time possibly due to measurement error. Work is underway to establish a gold standard assay.
3. Cellular senescence [Thesis: Cells secrete certain harmful factors; become resistant to apoptosis; may persist in tissues for years, interfering with tissue repair]
Evidence for link to aging:
Conditional upon which senescence phenotype you use. With some phenotypes, evidence seems pretty strong that senescent cells accumulate with age and that their removal reduces aging phenotypes. But senescence is highly heterogeneous depending on the senescence triggers and tissues in question, so quantifying it is complex. And importantly, no specific, unique diagnostic for senescence exists: some senescence biomarkers are present in situations that don’t fall under the umbrella of “cellular senescence”.
Available measures:
Largely because of the issues listed above, a gold standard measurement of cellular senescence that uniquely and universally identifies cellular senescence across tissues doesn’t exist. Many efforts to quantify senescence in biopsies from different human tissues are underway.
4. Epigenetics [Thesis in this context: that DNA methylation plays a role in aging and aging diseases. Site-specific percent methylation can be quantified in the form of an “epigenetic clock” that tracks chronological aging, risk of disease, or other metrics]
Evidence for link to aging:
The authors suggest that evidence for epigenetic clocks being good markers for aging is promising. They describe a recent generation of epigenetic clock that has been strongly predictive of mortality and a bunch of age-related adverse health outcomes, including disability and dementia.
They also discuss a novel DNA epigenetic modulator called 5hmC, or hydroxymethylcytosine, which has been associated with neurodegenerative diseases like Alzheimers, and with brain aging more generally.
Available measures:
DNA methylation is easily assessed in blood cells and tissues via microarrays, pyrosequencing, or whole-genome bisulfite sequencing methods. Unlike the other epigenetic operators histone modification and noncoding RNA, whose measurement is expensive, time consuming, and not fully standardized, DNA methylation is relatively stable over time.
The development of epigenetic clocks is based on an agnostic statistical approach because the biological mechanisms driving the clock are as yet unknown.
5. Mitochondrial dysfunction [Thesis: Accumulation of damage to the mitochondria and mitochondrial DNA induces aging because it reduces energy availability and/or increases reactive oxygen species or ROS production]
Evidence for link to aging:
The authors seem to consider mitochondrial function a potentially powerful biomarker for aging. Several different methods and studies have shown that the degree of oxidative phosphorylation declines with aging in various tissues like the heart and skeletal muscle. And reduced mitochondrial function is associated with mobility declines with age.
Available measures:
There are no measures of mitochondrial function in humans that are fully satisfactory.
Static measures like P31 MRS (Phosphorus 31 magnetic resonance spectroscopy), an in vivo method of studying mitochondrial function, are noninvasive and reliable, but too expensive for large population studies. P31 MRS only measures global skeletal muscle oxidative phosphorylation (this depends not just on mitochondrial function, but also on the capacity of the circulatory system to deliver enough oxygen to the mitochondria).
Muscle biopsies are invasive, but safe and enable a wide range of measurements and assays, many of which have been associated with aging and chronic conditions
The significance to aging of assays for measuring ROS, antioxidant defense, oxidative damage is uncertain, because they were usually studied in specific medical conditions rather than in the context of aging studies.
mtDNA copy number (number of mitochondrial genomes per cell) may provide information on mitochondrial physiology that is relevant for aging and age-related diseases. High copy number is considered a possible proxy measure of mitochondrial volume and function, and is associated with better health and survival among older persons. But this association may be reversed in diabetes.
The authors describe these measures as potentially powerful biomarkers of biological aging, but note that they require very careful standardization. Blood measurements may be affected by changes in circulating cells, for example. Also, a high mtDNA copy number, while associated with better health and survival among old people, may also indicate chronic tissue hypoxia.
6. Proteostasis [Thesis: Changes to the protein synthesis, folding, and degradation processes possibly accelerate aging]
Evidence for link to aging:
The authors seem relatively convinced by the evidence for a link between loss of proteostasis and aging. They talk about genetic variants in core autophagy genes contributing to human diseases like Parkinson’s disease, how autophagy becomes defective with aging and contributes to immunosenescence, and how compounds that modulate autophagy have shown anti-aging properties—things like spermidine, resveratrol, and urolithin A.
Available measures:
Measurement is challenging. Static measures of autophagosome accumulation (autophagosomes are vesicles that engulf cellular material to transport to lysosomes for degradation) are simple, but unreliable.
Measuring the dynamic flux of the autophagic process by quantifying accumulation of autophagosome cargo after inhibiting lysosomal proteolysis (cleaving proteins into smaller components) is better and more reliable.
But adequately quantifying autophagy requires multiple approaches. Most are expensive, labor-intensive, low-throughput. No high-throughput method for quantifying proteostasis exists.
There are a few new methods floating around, but they need further validation before they can be used in clinical studies.
7. Stem cell exhaustion, deregulated nutrient sensing, altered intercellular communication [Grouped together because unsuitable for inclusion]
Stem cell exhaustion may play a role in aging by interfering with self-renewal of differentiated cells in tissues and organs. Limited data.
Nutrient sensing may be relevant to aging in the context of caloric restriction, though it is still not totally clear whether caloric restriction truly impacts aging in humans. Boundaries of this hallmark not well defined.
Intercellular communication is such a vague term that it basically encompasses almost all known physiological mechanisms, so can’t really say much about the impact on aging.
Discussion notes
- The authors suggest that the measures with the strongest links to aging + greatest measurement sophistication are DNA methylation and mitochondrial function, followed by cellular senescence and autophagy (proteostasis).
- The authors’ “final conclusions” for each aging biology measure don’t seem particularly consistent. For example, genomic instability and telomere length seem to have similar levels of evidence for links to aging, but the authors describe genomic instability as “likely important” to biological aging while telomere length importance is “unclear”. Unclear what level of evidence the authors’ require to reach conclusions about the [non]importance of a given mechanism to aging.
- The authors point out that something common seems to happen downstream of all these processes: inflammation. Every measure described in this paper directly or indirectly causes an inflammatory state.
- A challenge of focusing on hallmarks or measures such as these: these hallmarks aren’t “models of aging”—that is, they don’t describe a system of things interacting to age a human—so “measuring a hallmark” isn’t automatically measuring a process that drives aging.
- Q: Is there possibly something upstream driving all of the measures described in this paper? A: Aging is more likely to occur in the system of informational organization and communication between components, e.g. misfolded proteins, senescent cells, DNA damage, rather than in the components themselves. If we were to sort out the components into piles, we would no longer have life and thus no longer have aging. The system is likely not linear, but rather a connected web in which some nodes have consistent strong effects on “downstream” nodes even as those nodes are directly or indirectly affected by those downstream nodes.
- Over-reliance on established “hallmarks” of aging may make us miss other mechanisms important to aging, e.g. adaptive immunity, ECM signaling, inflammation, possibly circadian rhythms, chemical modifications outside cells.
- We are still far from clinical measurements for most “hallmarks” of aging, partly because many hallmarks, e.g. senescent cells, are not sufficiently defined.
Statistics of Biology

No paper
Journal Club Presenter: Lada Nuzhna
Motivation for including: Having gone through a range of promising data from model organisms, we know that aging can be modulated by interventions. Has there been any progress in human studies?
Slides: Link
Presentation date: 3/06/2022
Discussion notes
Longevity field is notorious for “everything is correlated with everything”. Things to be careful about in such papers:
Experiment & Control group: Lifespan studies are only as good as their controls. Remember the UW vs NIA studies on caloric restriction in monkeys? One showed lifespan extension and the other one didn’t. It shows the importance of getting your controls right. See also exhaustive thread Matt Kaeberlein had about this on twitter:
“ Short-lived controls are a major source of false-positive results in mouse lifespan studies. Metformin, nicotinamide riboside, intermittent fasting all suffer from this flaw. This graphic illustrates the problem”

In any longitudinal study: control for time. In any other study: think of anything that can impact your final outcome and control for it (age, baseline measure, gender, genetic composition)
Absence of control is unlikely to be a problem (it is a normal practice in science to include them!) - it is inadequate control that is more problematic:
No “placebo” interventions in control / Subjects aren’t blinded
Control group with different baseline measure
Small control groups
Controlled manipulation isn’t identical to experimental manipulation
The only difference should be in controlled variable!
Comparing 2 effects: to compare 2 effects, you need to make a direct comparison, not just the difference in their comparison to control. The only conclusion you can draw from comparing intervention to control is that it is different from control (or similar to control). The difference between 2 effects can be insignificant even if they seem different when you compare them to control
P-hacking: adding covariates, excluding subjects, switching outcome parameters, until a “significant” result is reached.
That’s how probability works: the more tests you run, the more likely you are to get a false positive. It is almost impossible to detect p-hacking if you are just reading a paper. A few ways one might watch for this:
distinguish pre-planned and exploratory data analysis
a p-hacked p-curve will have an overabundance of p-values just below 0.05
if the choice of analysis feels weird, then its most likely p-hacking
way to avoid it in clinical trials: pre-registration of your trial design
Summary of longevity intervention data in humans

No single paper
Journal Club Presenter: Tara Mei
Motivation for including: Having gone through a range of promising data from model organisms, we know that aging can be modulated by interventions. Has there been any progress in human studies?
Slides: Link
Presentation date: 5/22/2022
Summary
Rigorous studies of longevity interventions in humans are sparse. These notes cover:
Caloric restriction
Metformin
Parabiosis
1. Caloric restriction (CR)
Comprehensive Assessment of Long term Effects of Reducing Intake of Energy (CALERIE): What are the effects of 2 years of caloric restriction on cardiovascular mortality and morbidity in humans?
Methods:
- Participants reduce caloric intake by 25% below baseline over a 2-year period (while 25% was the intended magnitude of CR, participants did not actually achieve this amount of CR)
- N = 218: 21-50 y.o. men, 21-47 y.o. women
- BMI 22.0-28.0
- Everyone gets a daily multivitamin and mineral supplement and calcium supplement
- Endpoints and schedule of outcome evaluations here
Results:
- Decreased total cholesterol, triglycerides, insulin resistance, mean blood pressure, some inflammation biomarkers
- Improved several liver biomarkers
- Weight loss, mostly from fat loss
- No significant effect on glucose measures, IL6, TNF-α, norepinephrine
- Adverse effects: In CR group, decrease in bone mineral density, increase in reproductive disorders and skin disorders. No effect on depression, verbal memory, sexual function, perceived hunger, eating disorder pathology.
Notes:
- Mean BMI was 25.1 (control group) and 25.2 (CR group) (BMI ≥ 25 is classified as overweight)
- Mean body fat percentage was 33.6 (control group) and 32.9 (CR group) (body fat percentages ≥ 32 and ≥ 26 are classified as overweight for women and men respectively)
- The percentage of participants who experienced certain adverse effects (e.g. nervous system disorders) increased in the CR-normal weight group, but decreased in the CR-overweight group compared to controls
- Why didn’t the study include participants with BMI < 22? Possibilities: safety—adverse side effects in lower-BMI participants could limit the whole study; less feasible to maintain 25% CR in lower-BMI participants
- What is the right CR for a given individual, given genetic interactions?
- Given that compliance with 25% CR is so hard (and wasn’t achieved in this case!), is the time and money spent on human CR studies well spent?
- Intermittent fasting (limited feeding windows so that participants spend longer periods without food) seems to have some benefits for age-related health conditions such as diabetes mellitus, cardiovascular disease, and cancers. However, 1. we do not fully understand the mechanisms involved, 2. we are unsure about the degree to which adherence to an intermittent fasting regime is practical, 3. research participants have mostly been overweight, young and middle-age adults, so it is unclear how intermittent fasting might affect other groups, 4. other evidence suggests no cardiometabolic benefits to intermittent fasting
- The golden unanswered question: Do the effects of CR come from lower calorie consumption, or from the weight loss that accompanies lower calorie consumption?
2. Metformin
Metformin is a small molecule drug that typically treats Type 2 diabetes by lowering glucose levels without triggering hypoglycaemia. This drug is appealing as a potential longevity intervention: it is safe, orally administered, cheap, widely available, and has been around for a long time. Doctors have prescribed it to treat conditions other than Type 2 diabetes, including “prediabetes”, gestational diabetes, polycystic ovary syndrome, and weight gain from antipsychotics.
No one has completed clinical trials explicitly testing the efficacy of metformin on human longevity or aging diseases, so it is unclear whether metformin increases maximum human lifespan. The main current effort in this direction is the Targeting Aging with Metformin (TAME) trial, which is in the process of raising funds. There is, however, some evidence that metformin helps with aging diseases, e.g. this meta-analysis suggests that diabetics on metformin have a lower incidence of certain aging diseases than do other diabetics and/or non-diabetics.
Notes:
Are the health and mortality changes that we see in diabetics taking metformin or other drugs independent of those drugs’ effects on the diabetes itself? All-cause mortality is lower in diabetics taking metformin than in non-diabetics and diabetics taking non-metformin therapies. But in the case of cardiovascular disease, whose occurrence is lower in diabetics taking metformin than in diabetics taking non-metformin therapies, no studies compared diabetics taking metformin to non-diabetics.
Can we generalize the changes we observe in diabetics taking metformin to non-diabetics taking metformin?
Are studies making fair comparisons—for example, adjusting for age? “Controlled for age” can be worse than actual age-matching because it makes assumptions about linearity and may not include all confounders. Ask: “What are the ways this adjustment could have been inadequate?”
How can we correct for sponsor bias in reporting? [seeking additional sources of information?]
3. Parabiosis
PLasma for Alzheimer SymptoM Amelioration (PLASMA) Study: Is young plasma safe and tolerable in patients with mild to moderate Alzheimer’s disease dementia?
Some evidence suggests that young blood reverses brain aging in old mice. The PLASMA study was an exploratory trial on the road to testing whether the same could be true in humans.
Methods:
- 4 weekly infusions of either plasma from young male donors or saline (control)—>6-week washout—>4 weekly infusions of an alternate treatment
- N = 18, 50-90 y.o. men and women with mild to moderate Alzheimer’s disease dementia
- A study partner very familiar with patients would attend all patient visits to evaluate. Exploratory outcomes: a change in baseline for the Alzheimer Disease Assessment Scale-Cog (ADAS-Cog) 13-item version, Trail Making Test (TMT) parts A and B, Geriatric Depression Scale, Neuropsychiatric Inventory, Clinical Dementia Rating Scale Sum of Boxes, Functional Activities Questionnaire, and the Alzheimer Disease Cooperative Study Activities of Daily Living Inventory in mild cognitive impairment; fMRI analyses such as functional connectivity maps for left and right hippocampus regions of interest
Results:
- No serious adverse events
- Mild adverse events like hypertension, dizziness, headache, sinus bradycardia, sinus tachycardia
- No statistically significant differences in mood, global cognition, or functional connectivity; some improvements in functional abilities. But small sample size, changes in study design, and short treatment duration make these assessments purely exploratory.
4. Other stuff
- Many clinical trials of senolytic agents (drugs that selectively clear senescent cells) to treat e.g. osteoarthritis, Alzheimer’s disease are underway
- The Rejuvenation Roadmap compiles rejuvenation technologies currently in development. Many of the studies listed are small and/or poorly controlled.
- Rapamycin, an FDA-approved small molecule mTOR inhibitor, has demonstrated lifespan extension and reversal of age-related changes in several model organisms such as mice. Current efforts to study its potential role as a longevity intervention in humans include the University of Washington Rapamycin Study, which collects data on the health of people already taking low-dose rapamycin, and the Participatory Evaluation (of) Aging (With) Rapamycin (for) Longevity Study (PEARL), a clinical trial aimed at determining the efficacy of Rapamycin in improving health and reducing clinical aging measures in healthy older adults.